
| Home | Update News | Performance | Documentation | Download | Instructions | Contact |

Step 1: Indicate whether the query protein is deuterated Step 2: Fill in information about pH. By default, pH is set to 5. Step 3: Fill in informationa bout Temperature (K). By default, Temperature is set to 298K Step 4: Indicate whether the phosphorylated residue is calculated. The chain ID should be assinged in PDB ID input box or file name  Step 5:select the type (all, backbone or proton only) of chemical shift you want to predicted. Selecting "all" will predict both backbone and side-chain chemical shifts. 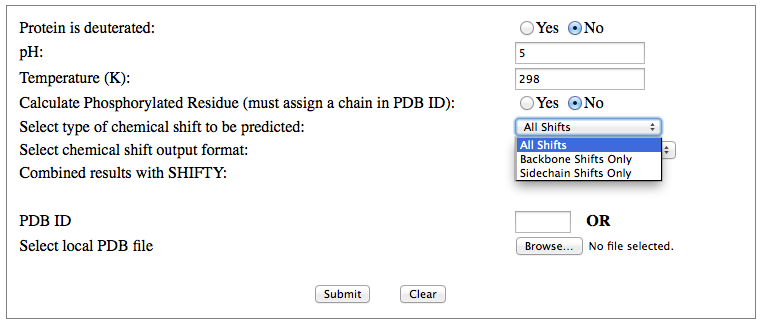 Step 6:Select the output format you desire (tabular, comma separated, NMR-STAR, or NEF format). By default the output format is "tabular". 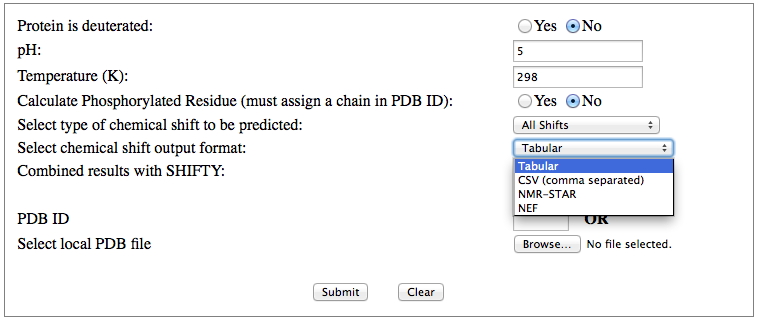 Step 7:Indicate whether to combine results from sequence-based method (SHIFTY). If this option is check, SHIFTX2 will combine the results from both sequence-based and structure-based prediction methods. Step 8: Type in a valid PDB ID (if multiple chains are present, only the first chain will be predicted). You will have the option of selecting which chain to predict using the standalone version of SHIFTX2. 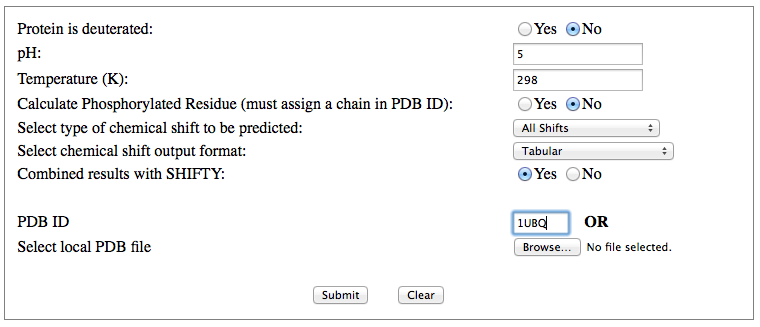 Step 9: You can upload a local PDB file from your computer for prediction. If this option is used, you may clear the PDB ID in the previous step. 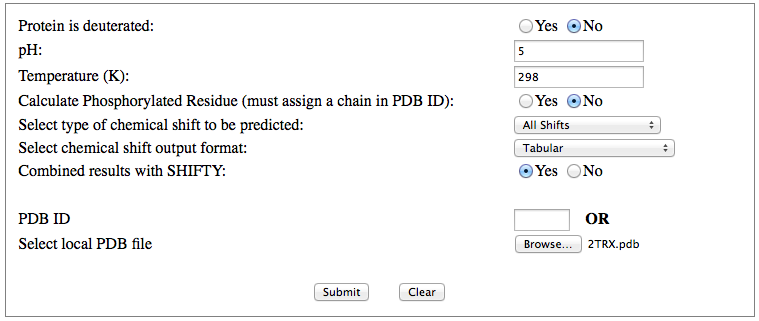 Step 10: Press the submit button to submit, or press the clear button to clear out the form for correction. Step 11: View the results. For example, the following output snippet is generated using PDB ID "1UBQ". The results are devided into 3 sections "backbone chemical shifts", "side-chain carbon shifts" and "side-chain proton shifts" 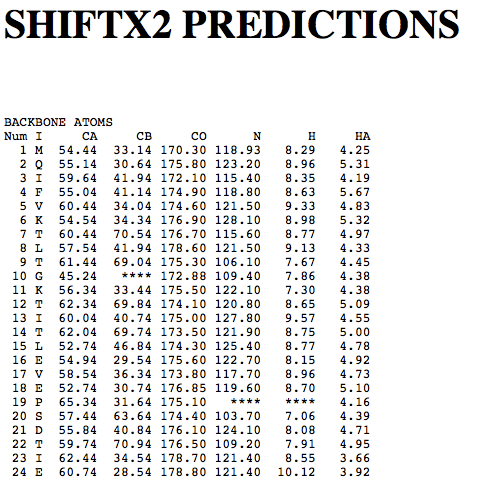 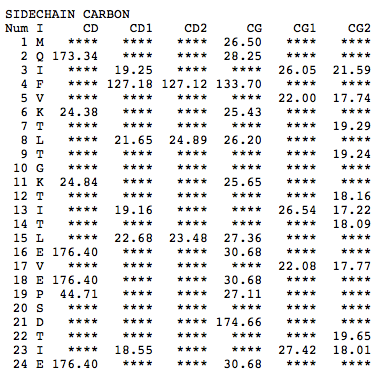 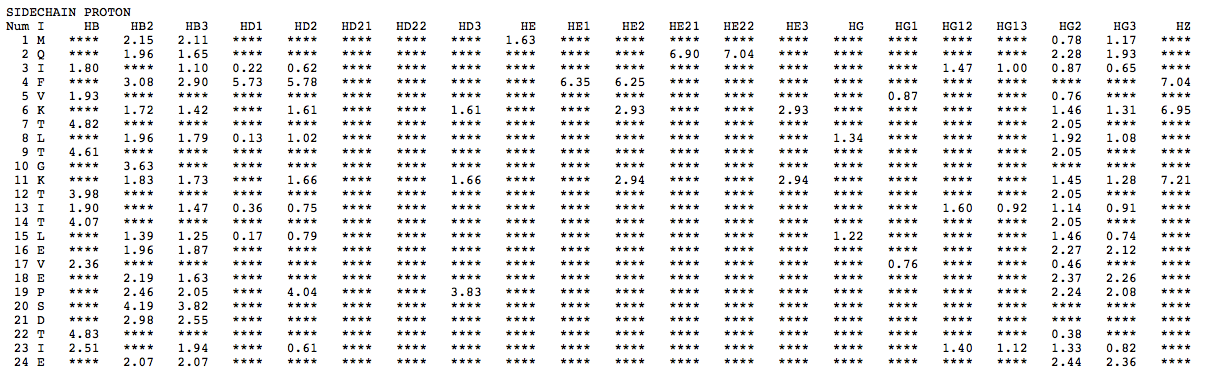 |